Les fabricants qui souhaitent commercialiser un dispositif médical aux Etats-Unis doivent satisfaire un ensemble d’exigences réglementaires. Parmi celles-ci, la usability (ou Human Factor Engineering) mérite d’être anticipée, car les efforts nécessaires pour répondre aux attentes de la FDA peuvent s’avérer conséquents.
La FDA requiert que les fabricants mettent en place une évaluation de validation (appelées Human Factor validation evaluation) avec la version finale du dispositif afin d’apporter la preuve que le dispositif peut être utilisé en toute sécurité par les futurs utilisateurs. De plus, afin de s’assurer que les participants aux tests soient bien représentatifs des futurs utilisateurs, la FDA demande à ce que les tests utilisateurs de validation soient faits sur le sol américain avec des résidents américains.
Cela implique donc de recruter au minimum 15 participants américains, voire plus si le dispositif est utilisé par plusieurs profils utilisateurs (dans ce cas il faudra prendre 15 utilisateurs pour chaque profils) et de mettre en place une méthodologie de tests utilisateurs aux Etats-Unis répondant aux exigences définies dans la guidance FDA (Applying human factors and usability engineering to medical devices, 2016).
Evidemment, le profil des participants doit également correspondre au profil des utilisateurs finaux et une justification de la sélection des profils doit être apportées. Par exemple, si le dispositif est utilisé par des infirmières et des chirurgiens et que ces deniers exécutent des tâches différentes avec le dispositif, il faudra recruter 30 professionnels de santé aux Etats-Unis.
De plus, il faudra éventuellement trouver une salle de tests utilisateurs représentative de l’environnement réel d’usage. Cela implique parfois de faire appel à des centres de simulation médicaux qui mettent à disposition des salles fictives mais réalistes de chambres d’hôpital, de bloc opératoire, etc.
Mettre en place une étude de cette envergure implique de bien planifier la usability dans le planning projet globale et de mettre en œuvre des efforts et des ressources qui ne sont pas négligeables.
La réponse rapide est non, pas toujours ! La FDA a précisé dans quels cas et comment justifier le fait de ne pas faire d’évaluation de validation. En effet, en janvier 2022, elle a publié une version préliminaire (draft) d’une guidance intitulée “Content of Human Factors Information in Medical Device Marketing Submissions”. Ce document vient préciser les attentes de la FDA en matière de facteurs humains et d’évaluation de l’ergonomie des dispositifs médicaux, notamment en ce qui concerne les situations dans lesquelles des sessions de tests utilisateurs peuvent ne pas être nécessaire.
Cette guidance draft s’inscrit dans la continuité de la guidance de 2016 et vient la compléter. Elle vise à renforcer la sécurité d’utilisation des dispositifs, tout en reconnaissant que certaines soumissions peuvent s’appuyer sur des données existantes plutôt que de revalider des interfaces déjà maîtrisées.
Cette approche, bien que plus souple, ne dispense pas les fabricants de leur responsabilité de démontrer la sécurité d’utilisation de leurs dispositifs. En revanche, elle peut accélérer les démarches réglementaires, réduire les coûts de développement, et éviter des tests redondants, à condition de bien maîtriser les exigences de la FDA.
Pour préciser les différents cas de figure possibles, la FDA fourni un schéma de prise de décision qui amène à trois catégories de soumission différentes (voir le schéma ci-dessous).
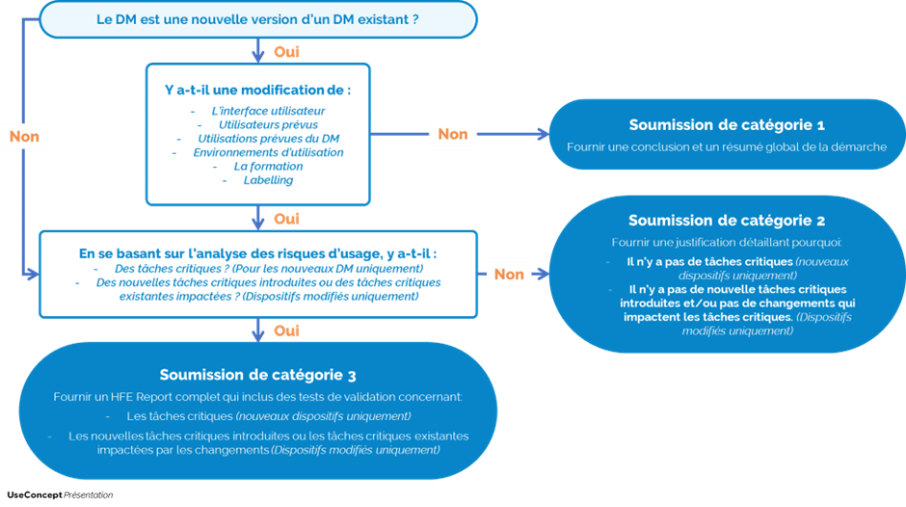
Ce type de soumission concerne les dispositifs qui ont déjà été commercialisés aux US par le fabricant. Dans ce cas-là, le fabricant a déjà soumis un dossier complet de HFE Report auprès de la FDA et il souhaite désormais développer une nouvelle version de son dispositif. Cependant, les modifications apportées sur le dispositif sont mineures, elles ne sont pas liées à des risques d’usage et peuvent même être transparentes pour l’utilisateur. Autrement dit, les modifications n’impactent aucun des éléments suivants :
➡️ L’interface utilisateur (à savoir le dispositif en lui-même, l’IFU, le packaging, etc.)
➡️ Les utilisateurs prévus
➡️ Les utilisations prévues
➡️ L’environnement d’utilisation
➡️ La formation
➡️ Le labelling
Exemple : Un fabricant développe un logiciel de détection des lésions gastro-intestinales. Le DM est déjà commercialisé et a déjà eu le FDA approval 510(k). Le fabricant à modifier l’algorithme de détection de lésion afin de l’améliorer. Les modifications ne changent pas l’interface utilisateur. Aucun test de validation supplémentaire n’est requis.
Ce type de soumission concerne deux cas de figures possibles :
☝️ Soit il s’agit d’une nouvelle version d’un dispositif déjà commercialisé auquel le fabricant apporte des modifications. Mais, ces modifications n’introduisent aucune nouvelle tâche critiques et n’impactent pas les tâches critiques précédemment identifiées. Dans ce cas-là, il faudra cependant apporter une justification en se basant sur l’analyse des risques d’usage du DM.
☝️ Soit il s’agit d’un nouveau dispositif qui n’a pas encore été commercialisé, mais qui n’est lié à aucune tâches critiques. Là encore, il va falloir apporter la preuve à la FDA que le DM est sûr à utiliser et qu’un travail d’identification des tâches critiques a été fait selon les attentes décrite dans la guidance.
Exemple : Un fabricant développe un oxymètre portable destiné à mesurer la saturation en oxygène chez des adultes, à domicile ou en établissements de santé. Le fabricant ne commercialise pas d’autre oxymètres, mais il utilise un predicate d’un autre fabricant. Le dispositif ne comprend aucune alarmes ou interprétation des niveaux d’oxygène et ne présente aucune tâche critique.
Ce dernier type de soumission concerne également deux types de situations :
1️⃣ Le dispositif est une nouvelle version d’un dispositif déjà commercialisé et les modifications faites impactent l’analyse des risques d’usage.
2️⃣ Le dispositif est nouveau, il ne s’agit pas d’une évolution d’un dispositif déjà commercialisé et son usage impliquent que les utilisateurs rencontrent des tâches critiques.
Dans cette catégorie de soumission le fabricant devra mettre en place des tests utilisateurs de validation aux US et fournir un dossier de HFE Report complet.
Exemple : Un fabricant développe une nouvelle version d’un appareil d’anesthésie qui régule la quantité de gaz anesthésiant. Le DM est déjà commercialisé aux US (510k approval). Le fabricant modifie entièrement l’interface graphique de l’écran tactile du DM et une nouvelle fonctionnalité est ajoutée. Le manuel utilisateur ainsi que les supports de formation, seront modifiés.
Evidemment, le fait de ne vouloir économiser du temps ou des ressources n’est pas une justification suffisante pour ne pas faire de tests de validation. Chez UseConcept, nous ne pouvons que vous conseiller de passer par l’étape des tests, car nous savons que c’est le meilleur moyen pour s’assurer qu’un dispositif est sûre d’usage et que son utilisation satisfait ses utilisateurs. Après avoir mis en place des sessions de tests formatifs ou de validation, nous savons que les fabricants sont à coups sûr, ravis de tous les précieux retours que les tests utilisateurs leur apportent.
Lorsque nous parlons de renoncer à un test de validation HF, nous ne vous conseillons pas de ne pas mettre de ne faire aucune des étapes du process usability décrit dans les normes. Nous vous conseillons d'effectuer le travail adéquat pour démontrer que votre produit est sûr et efficace et que les mesures d'atténuation que vous avez mises en place permettent de maitriser les risques d’usage.
La guidance précise que, dans tous les cas, une justification formelle et documentée est exigée. Cela passe notamment par la soumission d’un Human Factors Engineering Report (HFE Report)contenant :
Le but est de démontrer, de manière argumentée, que l’utilisateur pourra utiliser le dispositif en toute sécurité sans nouvelle validation.
La draft guidance FDA 2022 apporte une clarification bienvenue sur les cas où les tests utilisateurs peuvent être évités, en se fondant sur une approche basée sur les risques et la continuité des données. Pour les fabricants, c’est une opportunité de rationaliser leur stratégie de validation des facteurs humains, tout en restant conformes aux exigences de sécurité.

